PINNT is a support group for people receiving artificial nutrition. It is run by patients and was established over 25 years ago. PINNT is now a national charity providing mutual support and understanding to hundreds of adults and children and their families adapting to life on home artificial nutrition. As I receive both enteral and parenteral nutrition I joined the group and have found their advice very helpful, particularly when I was trying to arrange a holiday. You can find out more about PINNT by clicking HERE.
I wrote an article for the PINNT quarterly newsletter which was published last week. My aim was to raise awareness of Ehlers Danlos Syndrome among people receiving artificial nutrition. I received two emails from PINNT members within hours of it being published! You can read the article below…
As a child I was aware I was more flexible than most and oddly proud of it. Having fingers that bent 90 degrees backwards seemed kind of fun. But as a teenager I started getting injured easily, usually playing football. During one of many visits to casualty a doctor noticed how many previous x-rays I’d had, and asked me if I was being abused. I laughed, then assured him they were injuries from sport.
A few years later I returned to the same hospital with symptoms of fatigue, headaches, nausea, a pounding heartbeat and purple feet. I didn’t mention that I was very flexible or that I’d had lots of injuries. It didn’t occur to me such varied symptoms could be related. The doctor didn’t ask either. But the signs were all there in my medical history. If the doctor had asked the right questions and done a few simple tests, he could have diagnosed me with Ehlers-Danlos Syndrome, and advised me to keep as active as possible, then referred me to a specialist about my racing heart-rate. But that didn’t happen.
I fell ill in 1994, when there was a lot of research yet to be done and painful lessons to be learnt. The doctor asked me very few questions, and only did a couple of basic tests. Then he diagnosed me with M.E. (Chronic Fatigue Syndrome) and prescribed months of bedrest. I quickly got worse.
Finding information about medical conditions was difficult in 1994, as the internet was yet to take over our lives. But months later I read a newspaper article about a condition called Postural Orthostatic Tachycardia Syndrome (POTS) meaning a rapid heart beat while standing. The article described many of the symptoms I’d been struggling with, so I was immediately convinced I had the condition.
I had to wait eight months to be tested as there were only three units in the UK at the time that had the necessary equipment, but it finally confirmed I had POTS. I was referred to Professor Christopher Mathias, a neurovascular specialist, who prescribed a drug to raise my blood pressure and recommended a progressive exercise programme. I started to feel better and built up to walking more than ten miles a day. However due to my rapid heart-beat I still struggled with some basic tasks. Professor Mathias suggested I try more strenuous exercises, but I quickly developed muscles strains and joint pain that wouldn’t heal. This pattern carried on for several years.
In 2001 I went into hospital for some cardiovascular tests. The doctors noticed my body shape was unusual; I’m tall, with long arms, legs, fingers and toes. They measured my body as they thought my ‘marfanoid’ body shape might point to a connective tissue disorder. They speculated that the connective tissue disorder could be the cause of my POTS symptoms.
I was referred to Professor Rodney Grahame, a rheumatologist, who found most of my joints were ‘hypermobile’ (double jointed). He diagnosed me with Ehlers-Danlos Syndrome (Hypermobility type). EDS is a group of conditions caused by genetic alterations in collagen, Hypermobility type being the most common of the six forms. Collagen is a protein which gives strength to many vital body tissues, such as skin, tendons, ligaments and blood vessels. So EDS can cause very varied problems, from joint pain to fainting. I was the first patient Professor Mathias’s department had found with both EDS and POTS and they have since found 80-90% of POTS cases are caused by EDS!
In a healthy person, blood vessels constrict slightly on standing to help cope with gravity. In someone with EDS, the weak collagen in blood vessels causes them to expand under pressure rather than constrict, so blood sinks into the lower legs causing the symptoms of POTS. Unfortunately there is no treatment for EDS, so the breakthrough didn’t lead to any progress for me. Despite endless physiotherapy I’ve got significantly worse in the years since, in varied ways.
In 2010 I started getting tachycardia after eating. It was pretty difficult. Eating a sandwich became like an extreme sport for me, as it was followed by a rapid heart-rate that could last an hour. Digestive problems are common with EDS, and the issue got slowly worse until I could barely eat or drink at all. I spent nearly a year in hospital while doctors tried to find a solution and my weight fell to eight stone. I had a PEG tube (Percutaneous Endoscopic Gastrostomy tube) inserted into my stomach enabling liquid food to be pumped in. The tube was extended into my duodenum and numerous medications were tried, but I could only tolerate about 1400 calories a day.
Over the next couple of years my body showed signs of malnutrition and my weight fell to seven stone. My body mass index was 13.2, ‘Very Severely Underweight’. I was re-admitted into hospital where I began to be fed by parenteral nutrition. Intravenous feeding has obvious risks, but it was the only treatment option left for me.
I’ve been receiving parenteral nutrition for two years now and I’ve gained over three stone. My blood tests are almost normal after several years of problems, and I look and feel better too. I miss food every day, but I’m grateful for the extra time peg feeding and parenteral nutrition have given me. Without them, if nature had been left to run its course, I’d have died in 2010.
Thanks to the amazing medical care I’ve received in the last few years my situation is easier now and my health improving. However my case remains an example of what can happen if EDS isn’t diagnosed early and managed appropriately. It took three years for me to be treated for POTS, and seven years to be diagnosed with EDS. By this time I’d already developed a number of long term joint problems which still limit my mobility to this day, confining me to a wheelchair. Progress has been made since then, but lack of awareness of EDS among the public and even the medical community is still a big problem. But I’m optimistic. EDS may be rare, but the internet has united our community and we’re all working to increase awareness, raise funds, and encourage research towards a cure.
Each time I read one of your excellent blogs, I just hope that word will spread. The mistakes made in your life so far – in diagnosis and treatment, in particular – should never have happened and should now be impossible. Can we be so confident? Only I think when EDS and POTS are part of normal medical concerns, when everyone has heard of them and everyone unites in making sure that those with EDS get the early action they deserve.
Keep writing. One day, everyone will hear that “Who” that bothers Horton.
A Reluctant Contortionist
February 6, 2016 @
3:23 pm
Thankfully doctors did learn from my case, which helps me live with the lifelong consequences of those early mistakes. But unfortunately many people experienced a similar battle to be diagnosed as I did, and still do today.
EDS will always be slow to be recognised in some people as the symptoms can be so diverse. But thankfully awareness among both the public and medical profession is improving. There’s a lot more progress to be made, but diagnosis times have reduced and there’s research being done into the genetic cause of the condition and the best ways to treat it. – Thanks for your comment.

 PINNT is a support group for people receiving artificial nutrition. It is run by patients and was established over 25 years ago. PINNT is now a national charity providing mutual support and understanding to hundreds of adults and children and their families adapting to life on home artificial nutrition. As I receive both enteral and parenteral nutrition I joined the group and have found their advice very helpful, particularly when I was trying to arrange a holiday. You can find out more about PINNT by clicking HERE.
PINNT is a support group for people receiving artificial nutrition. It is run by patients and was established over 25 years ago. PINNT is now a national charity providing mutual support and understanding to hundreds of adults and children and their families adapting to life on home artificial nutrition. As I receive both enteral and parenteral nutrition I joined the group and have found their advice very helpful, particularly when I was trying to arrange a holiday. You can find out more about PINNT by clicking HERE.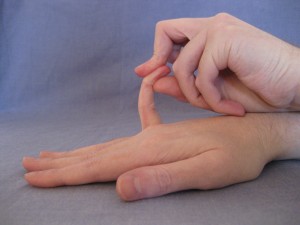 A few years later I returned to the same hospital with symptoms of fatigue, headaches, nausea, a pounding heartbeat and purple feet. I didn’t mention that I was very flexible or that I’d had lots of injuries. It didn’t occur to me such varied symptoms could be related. The doctor didn’t ask either. But the signs were all there in my medical history. If the doctor had asked the right questions and done a few simple tests, he could have diagnosed me with Ehlers-Danlos Syndrome, and advised me to keep as active as possible, then referred me to a specialist about my racing heart-rate. But that didn’t happen.
A few years later I returned to the same hospital with symptoms of fatigue, headaches, nausea, a pounding heartbeat and purple feet. I didn’t mention that I was very flexible or that I’d had lots of injuries. It didn’t occur to me such varied symptoms could be related. The doctor didn’t ask either. But the signs were all there in my medical history. If the doctor had asked the right questions and done a few simple tests, he could have diagnosed me with Ehlers-Danlos Syndrome, and advised me to keep as active as possible, then referred me to a specialist about my racing heart-rate. But that didn’t happen.
February 2, 2016 @ 6:35 pm
Each time I read one of your excellent blogs, I just hope that word will spread. The mistakes made in your life so far – in diagnosis and treatment, in particular – should never have happened and should now be impossible. Can we be so confident? Only I think when EDS and POTS are part of normal medical concerns, when everyone has heard of them and everyone unites in making sure that those with EDS get the early action they deserve.
Keep writing. One day, everyone will hear that “Who” that bothers Horton.
February 6, 2016 @ 3:23 pm
Thankfully doctors did learn from my case, which helps me live with the lifelong consequences of those early mistakes. But unfortunately many people experienced a similar battle to be diagnosed as I did, and still do today.
EDS will always be slow to be recognised in some people as the symptoms can be so diverse. But thankfully awareness among both the public and medical profession is improving. There’s a lot more progress to be made, but diagnosis times have reduced and there’s research being done into the genetic cause of the condition and the best ways to treat it. – Thanks for your comment.